Here is a story of a pharmacist fighting for her kids battling cystic fibrosis, a disease that impacts the lungs, and liver causing CFLD, cystic fibrosis liver disease, pancreas causing CFRD, cystic fibrosis-related diabetes, gut-causing leaky gut, and dysbiosis. How to diagnose, and treat cystic fibrosis, the life expectancy in cystic fibrosis, how pharmacogenomic and nutrigenomic testing helps and the functional medicine approach to manage CF.
What is Cystic Fibrosis?
Cystic Fibrosis (CF) is a progressive, life-shortening genetic disorder that affects two of my three children.
According to the Cystic Fibrosis Foundation, nearly 40,000 children and adults are living with cystic fibrosis in the United States (an estimated 105,000 people have been diagnosed with CF across 94 countries).
A defective CFTR gene causes secretions (mucus) to become sticky and thick.
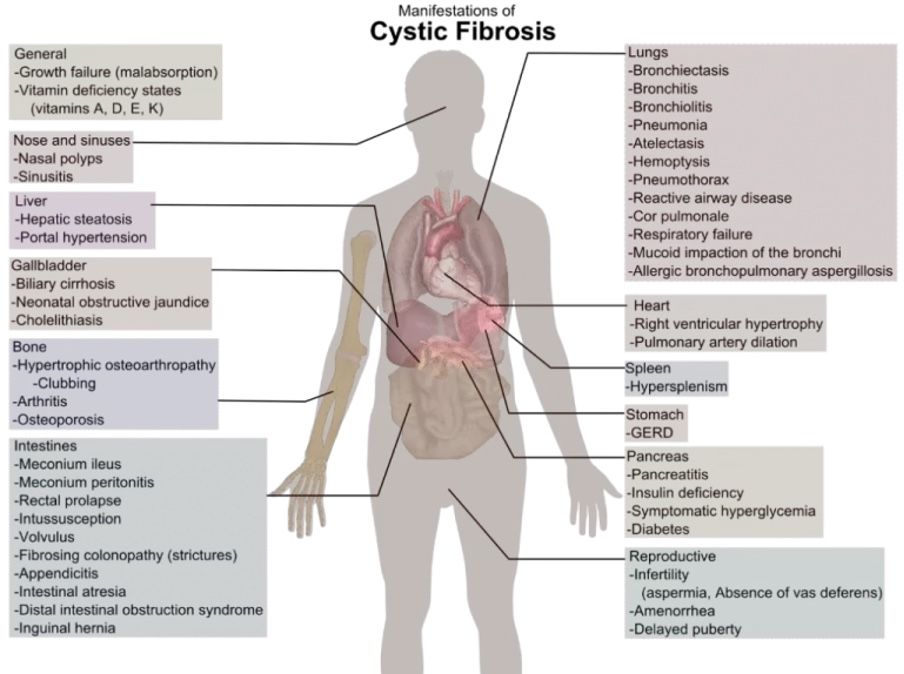
Impact of Cystic Fibrosis
Impact on Lungs
Thick mucus clogs the airways and traps bacteria.
CF is a vicious cycle of chronic lung infections, inflammation, and respiratory failure leading to lung transplants.
Not only the lungs, but CF can also impact the liver, pancreas, and gut causing cystic fibrosis liver disease, CFLD, and cystic fibrosis-related diabetes, CFRD, and CF gut respectively as described below.
Impact on Liver – CFLD
Cystic fibrosis-associated liver disease (CFLD) affects approximately 30% of patients, including my seven-year-old son (1).
CFLD, the cystic fibrosis liver disease is now considered the third cause of death after lung disease and transplant complications (1).
Diagnosing CFLD is a challenge. Most liver manifestations are only evident with the development of cirrhosis and portal hypertension.
Currently, there is no effective treatment for CFLD. The only treatment is preventing complications related to CFLD.
Impact on Pancreas – CFRD
Cystic fibrosis-related diabetes (CFRD) is one of the most common co-morbidity with CF. 85% of people with CF have pancreatic insufficiency (2). This can cause CFRD. The incidence of CFRD increases with age.
Thick mucus blocks pancreatic enzymes from entering the small intestine leading to malabsorption and maldigestion of nutrients and fat-soluble vitamins causing CFRD.
The digestive tract is forced to pass partially undigested food.
Therefore, pancreatic enzyme replacement therapy (PERT) is a staple of therapy in most people with CF and starts at birth to prevent CFRD.
Enzymes, due to mucus blockage, build up within the pancreas. Inflammation and scarring of the pancreas lead to insulin deficiency in CFRD similar to that of a person with type 1 diabetes.
Complications are not the same; therefore, this is referred to as Cystic Fibrosis related diabetes (CFRD).
Elevated blood glucose levels are detected well before the diagnosis of CFRD.
CFRD is associated with declining lung function, hence it is important to manage blood sugar levels with proper treatment.
CFRD sometimes shares the symptoms of both type 1 and type 2 diabetes still different than both. It can be distinguished from type 1 diabetes by its gradual onset. Also, insulin resistance seen in type 2 diabetes is only transient in CFRD.
Impact on the Gut – CF Gut
Intestinal malabsorption is severe and of early onset in virtually all people with CF (8).
The same pathophysiologic triad of obstruction, infection, and inflammation that causes disease in the airways also causes disease in the intestines (3).
This triad is called the “CF gut” (3).
The CF gut is associated with
- decreased motility
- increased intestinal permeability – leaky gut (4)
- dysbiosis – an imbalance of good and bad bacteria (6)
Who gets CF?
To have CF, you must inherit two copies of the defective CFTR gene, one from each parent.
My husband, middle child, and I only have one copy of the defective CFTR gene, so we are “carriers” but do not have the disease.
| Racial or Ethnic Group | Carrier rate (5) |
|
Caucasian, Ashkenazi Jews |
1 in 29 |
|
Hispanic American |
1 in 46 |
|
African American |
1 in 65 |
|
Asian American |
1 in 90 |
When parents are “carriers”, there is a 25 percent chance their children will have CF.
How do you treat Cystic Fibrosis?
Daily care is a full-time job in and of itself.
Airway clearance is performed manually (by hand) or by machine, depending on the age and size of the child.
This treatment is done a minimum of two times daily (30 minutes per session) and more frequently when sick.
During airway clearance, medications are administered via a nebulizer. These medications open airways, and thin mucus and treat infections when necessary.
Pancreatic enzymes are needed before every meal and snack so the body can digest.
It is not unusual for people to take 40+ pills daily, and I am being conservative here.
Other medications include but are not limited to fat-soluble vitamins, oral and IV antibiotics, inhalers, pain medications, nasal rinses, proton pump inhibitors, steroids, and modulators.
Modulators are a new class of drugs that target the underlying cause.
They are not a cure, but for some people, they feel like one.
Sadly, some people can’t tolerate them.
Modulators have given some people back the ability to breathe but at a tremendous cost.
People are making impossible choices between breathing and living with life-altering side effects.
My children have been blessed and have not experienced any side effects, and I hope that continues.
But let’s not forget that 5 -10 percent of the CF population will not be able to benefit solely from modulators or lack access to them.
We must continue research and outreach to help these people.
How do you diagnose CF?
Testing for cystic fibrosis –
- Sweat chloride test (cystic fibrosis sweat test)
- Newborn screening
- Genetic/carrier test
- Evaluation at accredited CF center
Life Expectancy in Cystic fibrosis?
Over the last few decades, life expectancy in cystic fibrosis has increased exponentially from a mere couple of years to 50.
Today, the median predicted age of survival is 34.1years.
We have made significant progress but still have a long road ahead.
My journey
During the pandemic, I decided to step away from working in a retail pharmacy setting to prevent exposing my high-risk kids.
I started researching ways to support liver health since my seven-year-old has CFLD.
In an instant, I fell down the rabbit hole of Functional Medicine (FM) that was introduced to me a few years prior by our nanny.
FM asks why symptoms are happening and then targets the root cause rather than naming the symptom and treating it with medications that don’t treat the underlying cause.
Imagine this…your bathroom sink is overflowing; you don’t reach for the mop to soak up the overflow, you turn off the tap. The water overflowing represents your symptoms, but the tap is causing the overflow and therefore is the root cause that needs to be addressed.

Pharmacogenomics
Pharmacogenomics (PGx) studies how your genes metabolize medications.
Testing provides information about a patient’s likelihood of having an adverse and/or therapeutic response to a medication.
We all metabolize medications differently.
There are guidelines dependent on whether you are a slow, normal, or rapid metabolizer.
Not only does PGx guide in dosing but on what medications to avoid.
Providers can use this as a tool to make precision-guided clinical decisions if prescriptions are needed.
Medication classes used in CF with actionable PGx guidelines
- Antibiotics
- Antidepressants/anxiety
- Antifungals
- Nausea
- ADHD/ADD
- Proton pump inhibitors
- Pain management
- Post-transplant medications
Pharmacogenomic testing decreases –
- Medications needed
- Adverse drug reactions
- Doctor visits/hospitalizations/ER visits
- Time to achieve therapeutic blood levels
- Cost by finding the right medicine the first time. No trial and error.
Pharmacogenomic testing increases –
- Efficacy of mental health medications
- Patient’s adherence to medications
- Bond between provider and patient
- Patient’s feeling of empowerment
PGx testing can be done from home and is a non-invasive test using DNA from a cheek swab.
Nutrigenomic testing
It is hard for people with CF to absorb fats and nutrients like B12 and beta-carotene.
The prescribed cystic fibrosis diet is the opposite of what you think of as a healthy diet. It is high calorie, high fat, high protein, and high sodium.
We know genes affect how the body responds to food and specific nutrients. And, in the other direction, the foods you eat and supplements you take can affect how your genes are expressed.
Gene variants and diseases like CF can alter how well a person can digest, absorb, metabolize, and synthesize certain nutrients.
Examples of how we can personalize nutrient requirements –
B12 impaired absorption in CF (9)
B12 is a vital nutrient that supports red blood cell production, energy production, nerve health, and DNA production.
Specific genetic variants can cause a lower capacity to absorb, utilize and/or transport vitamin B12 to parts of the body that need it.
It is possible to have a serum B12 “within normal limits” or “elevated” when you are, in fact, deficient.
The B12 is circulating in the blood and not being taken up into the tissues where it needs to be.
Beta carotene (10)
Beta carotene is a red-orange pigment found in plants and fruits.
The human body converts beta carotene into vitamin A, the bioavailable form our bodies can use.
Some people genetically can’t convert it and therefore require higher amounts of Vitamin A in their diet or may benefit from a supplement.
Functional Medicine approach to “CF Gut”
“All disease begins in the gut”
– Hippocrates nearly 2500 years ago.
The gut microbiome refers to all of the microbes in your intestines, which act as another organ that’s crucial for your health.
Why is the gut important?
- 95% of serotonin is made here
- Gut-brain connection through microbiome axis (11)
- Metabolic function is comparable to the liver
- Largest immune organ – 75% of the immune system sits here
CF is a non-modifiable risk factor for poor gut health. Left untreated will lead to systemic effects like depression, brain fog, fatigue, immune system dysfunction, joint pain, and autoimmune diseases.
The Functional Medicine model has a step-by-step process of evaluating, treating, and maintaining gut health, emphasizing diet and lifestyle.
We know bacterial overgrowth, dysbiosis, inflammation, increased intestinal permeability (leaky gut), and dysmotility occur in the Cystic Fibrosis intestine (12).
A leaky gut is when the intestinal junctions are no longer tight, allowing partially digested food, toxins, and bugs to penetrate the tissues beneath it.
This triggers additional inflammation and alters the gut microbiome.
Following contribute to dysbiosis and leaky gut –
- Medications
- Infection/Illness
- Chronic intestinal inflammation
- Surgery/trauma
- Nutrient deficiency
- Toxins
- Food reactions (allergies, sensitivities, or intolerances)
- Stress
Up to 40 percent of people with CF have small intestinal bacterial overgrowth (SIBO) (13).
Furthermore, CF patients are predisposed to a 5–10 times greater risk of colorectal cancer than the general population (14).
We need to focus on supporting gut health in people with Cystic Fibrosis!!!
Takeaways
- Functional Medicine needs to be added and integrated into the standard of care for Cystic Fibrosis.
- Pharmacogenomics helps ensure CF patients are getting the right drug, at the right dose, the first time, No more trial and error!
- Nutrigenomics identifies genetic predispositions to nutritional deficiencies in this population of people at increased risk.
- Immune function is vital in CF. Treating the gut will improve this system along with depression, anxiety, brain fog, pain, and fatigue.
Sources
- Kobelska-Dubiel N, Klincewicz B, Cichy W. Liver disease in cystic fibrosis. Prz Gastroenterol. 2014;9(3):136-141. doi:10.5114/pg.2014.43574
- Ghodeif AO, Azer SA. Pancreatic Insufficiency. In: StatPearls. Treasure Island (FL): StatPearls Publishing; May 1, 2022.
- De Lisle RC, Borowitz D. The cystic fibrosis intestine. Cold Spring Harb Perspect Med. 2013 Sep 1;3(9):a009753. doi: 10.1101/cshperspect.a009753. PMID: 23788646; PMCID: PMC3753720.
- De Lisle RC, Mueller R, Boyd M. Impaired mucosal barrier function in the small intestine of the cystic fibrosis mouse. J Pediatr Gastroenterol Nutr. 2011 Oct;53(4):371-9. doi: 10.1097/MPG.0b013e318219c397. PMID: 21970994; PMCID: PMC3188387.
- Zvereff, V., Faruki, H., Edwards, M. et al. Cystic fibrosis carrier screening in a North American population. Genet Med 16, 539–546 (2014). https://doi.org/10.1038/gim.2013.188
- Thavamani A, Salem I, Sferra TJ, Sankararaman S. Impact of Altered Gut Microbiota and Its Metabolites in Cystic Fibrosis. Metabolites. 2021 Feb 22;11(2):123. doi: 10.3390/metabo11020123. PMID: 33671639; PMCID: PMC7926988.
- Kliegman, Robert; Richard M Kliegman (2006) Nelson essentials of pediatrics, St. Louis, Mo: Elsevier Saunders ISBN: 0-8089-2325-0.
- Littlewood JM, Wolfe SP. Control of malabsorption in cystic fibrosis [published correction appears in Paediatr Drugs 2000 Jul-Aug;2(4):252]. Paediatr Drugs. 2000;2(3):205-222. doi:10.2165/00128072-200002030-00005
- Lindemans J, Neijens HJ, Kerrebijn KF, Abels J. Vitamin B12 absorption in cystic fibrosis. Acta Paediatr Scand. 1984;73(4):537-540. doi:10.1111/j.1651-2227.1984.tb09967.x
- Rust P, Eichler I, Renner S, Elmadfa I. Long-term oral beta-carotene supplementation in patients with cystic fibrosis – effects on antioxidative status and pulmonary function. Ann Nutr Metab. 2000;44(1):30-37. doi:10.1159/000012818
- Martin CR, Osadchiy V, Kalani A, Mayer EA. The Brain-Gut-Microbiome Axis. Cell Mol Gastroenterol Hepatol. 2018 Apr 12;6(2):133-148. doi: 10.1016/j.jcmgh.2018.04.003. PMID: 30023410; PMCID: PMC6047317.
- Jill Dorsey, Tanja Gonska,Bacterial overgrowth, dysbiosis, inflammation, and dysmotility in the Cystic Fibrosis intestine,Journal of Cystic Fibrosis,Volume 16, Supplement 2,2017,Pages S14-S23,
- Patel D, Shan A, Mathews S, Sathe M. Understanding Cystic Fibrosis Comorbidities and Their Impact on Nutritional Management. Nutrients. 2022 Feb 28;14(5):1028. doi: 10.3390/nu14051028. PMID: 35268004; PMCID: PMC8912424.
- Hadjiliadis D, Khoruts A, Zauber AG, et al. Cystic Fibrosis Colorectal Cancer Screening Consensus Recommendations. Gastroenterology. 2018;154(3):736-745.e14. doi:10.1053/j.gastro.2017.12.012

Dr. Julie Wharam is a Doctor of Pharmacy certified in pharmacogenomics. She founded Do Life Health, personalized precision medicine. She looks forward to launching her CF podcast and bringing precision medicine into the standard of care. Feel free to reach out to her at juliewharam@gmail.com, or join her FB CF support grouphttps://www.facebook.com/ groups/cysticfibrosishealth.